

DevelopingBiosimilars
© 2020, 2023 Amgen Inc. All rights reserved. USA-CBU-80774





















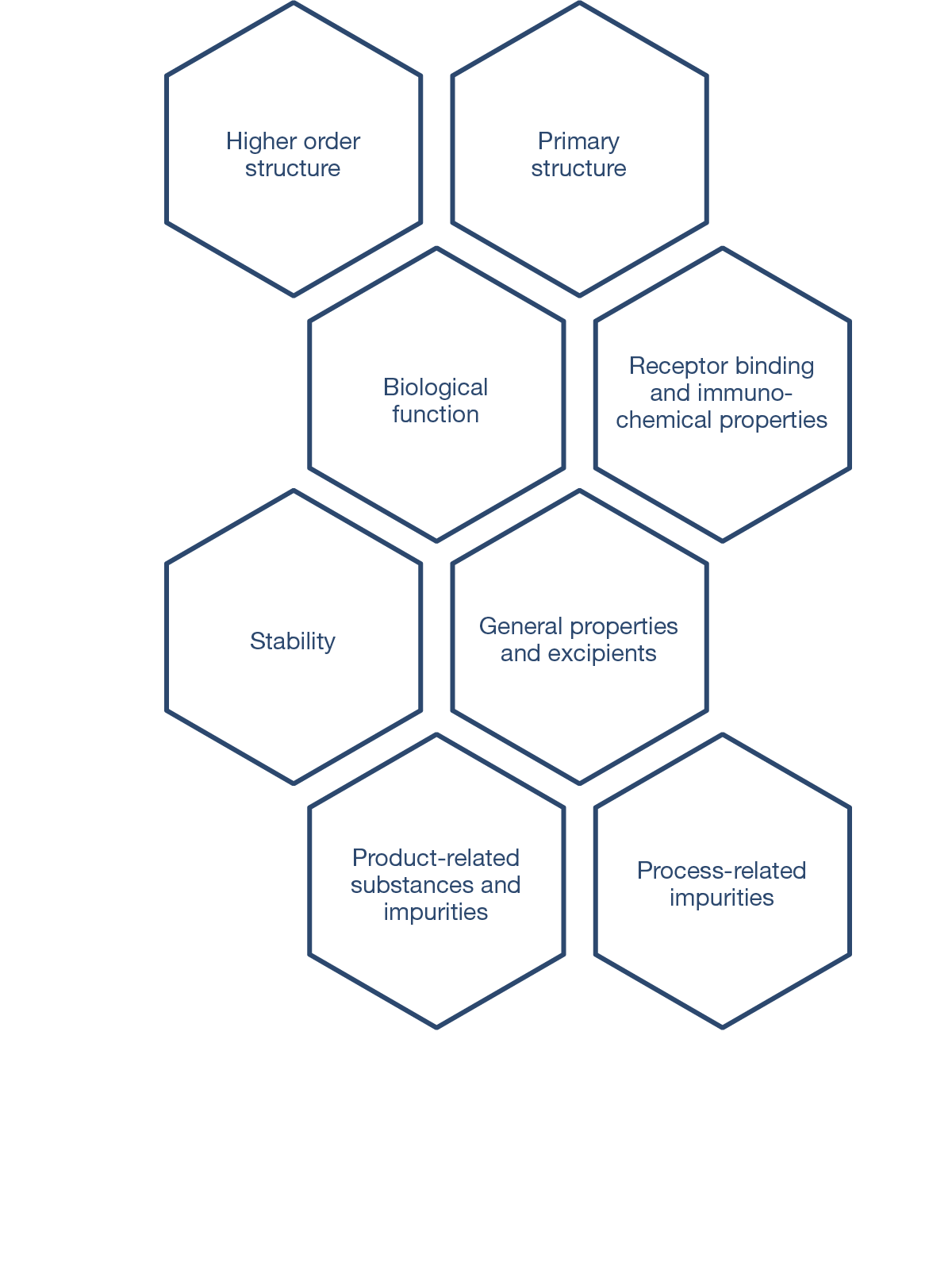
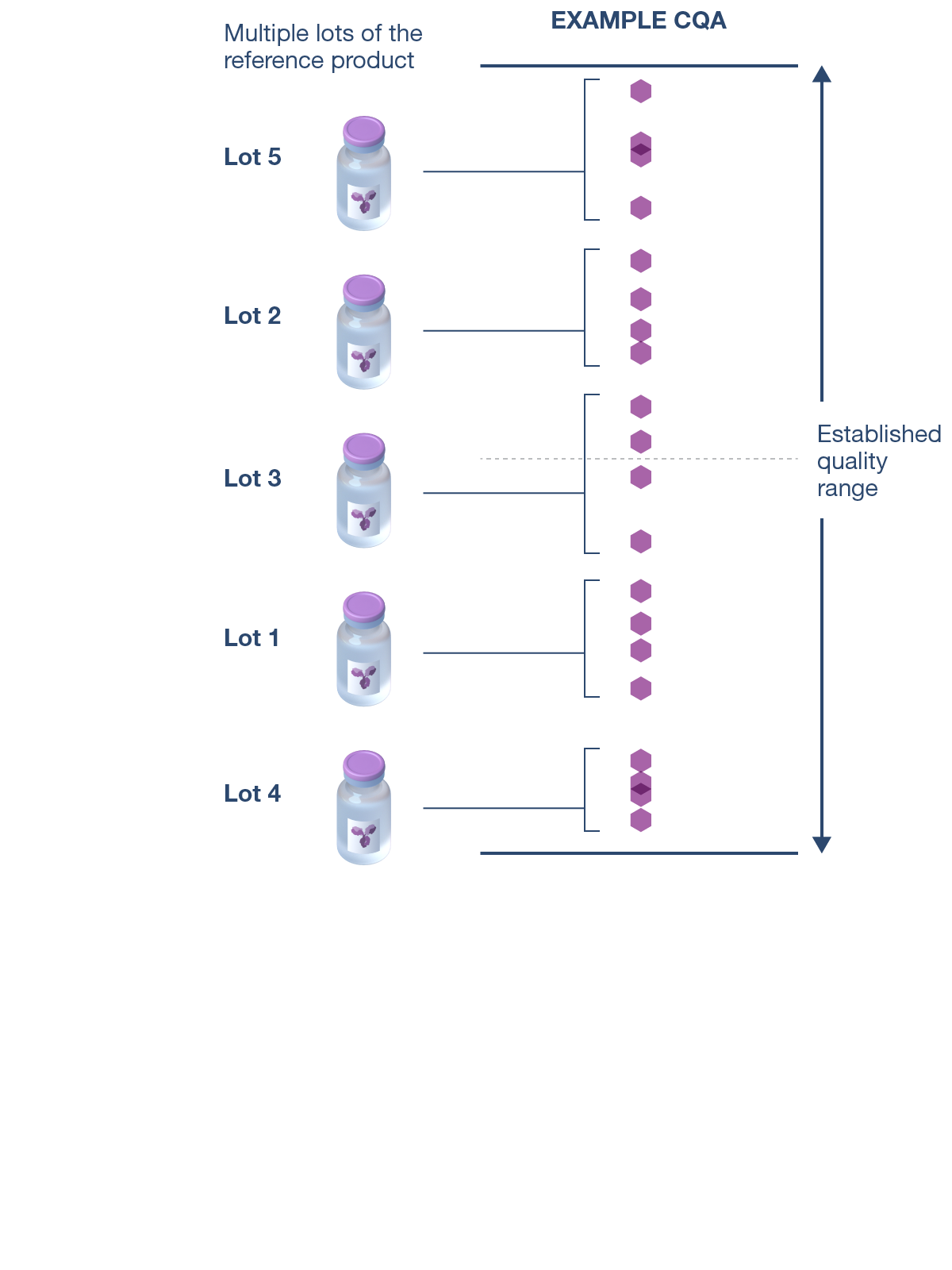
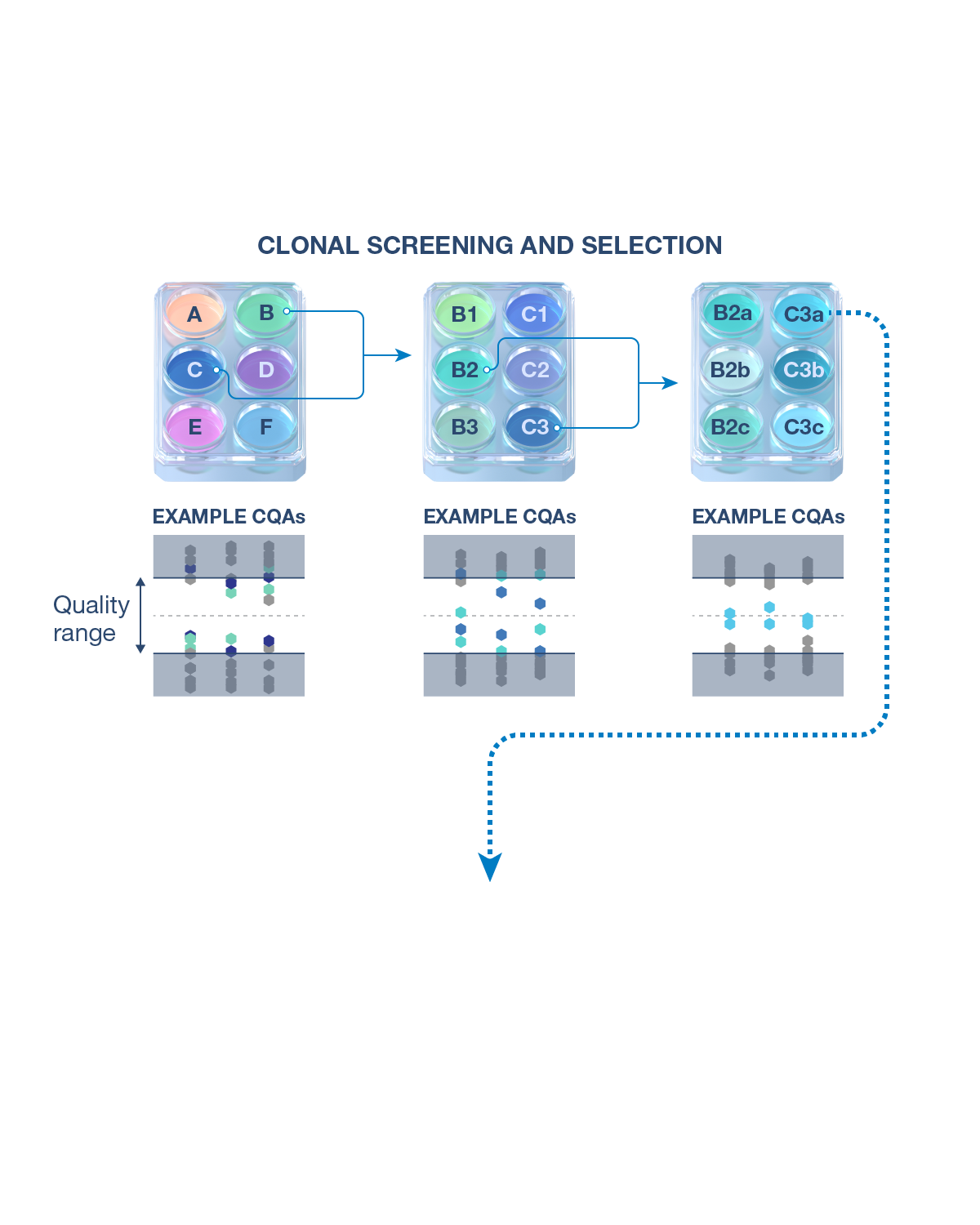
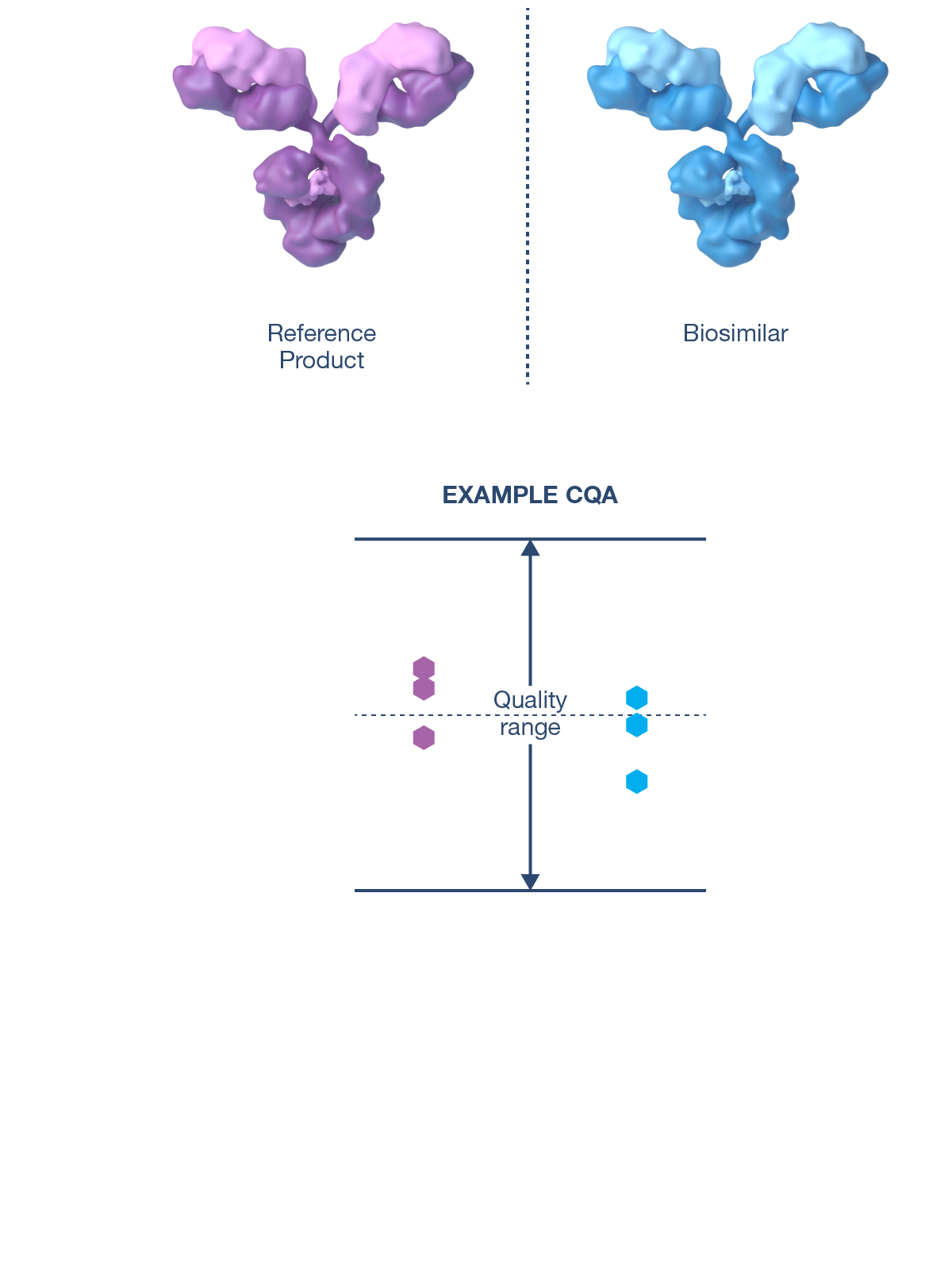
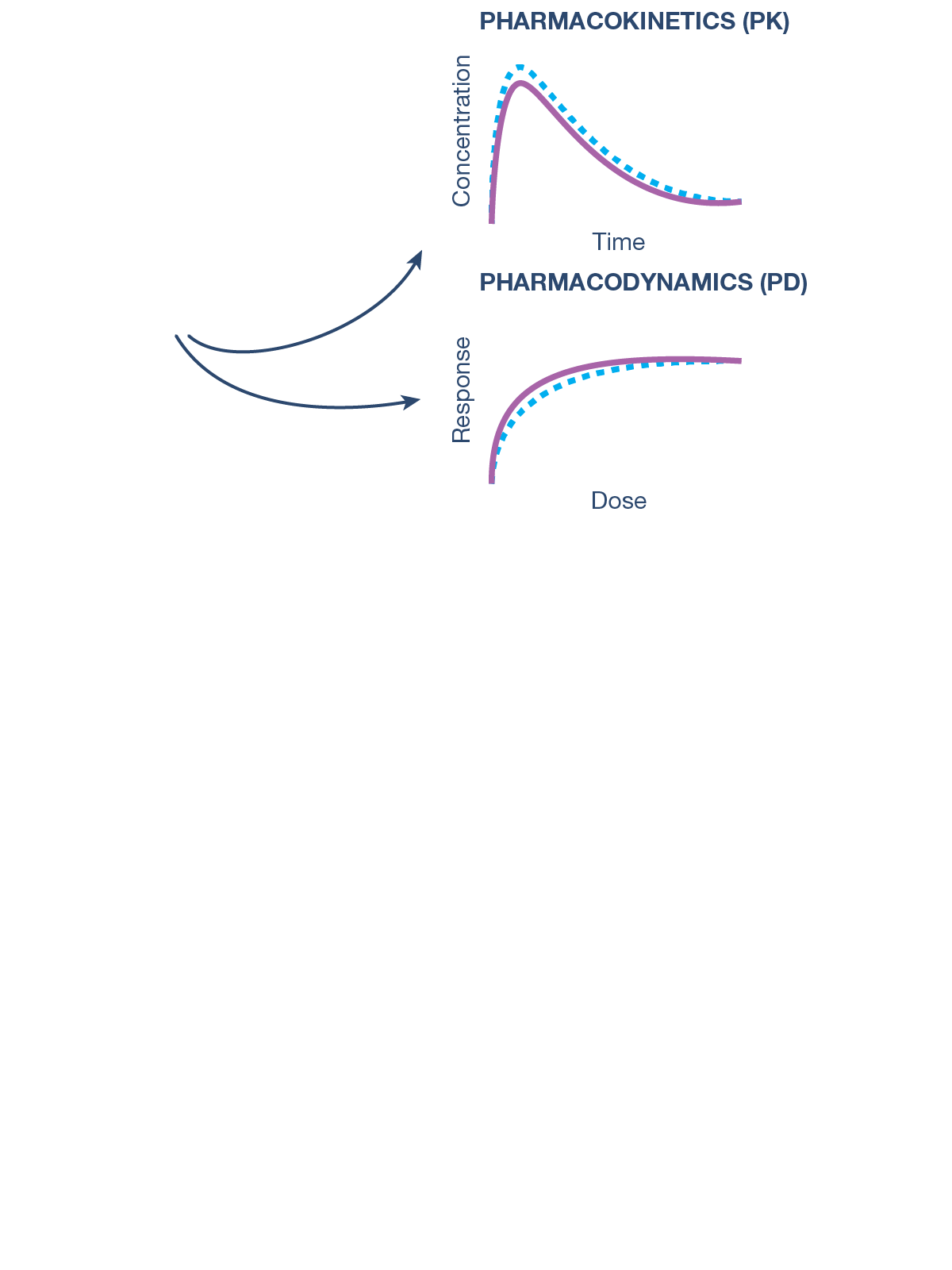
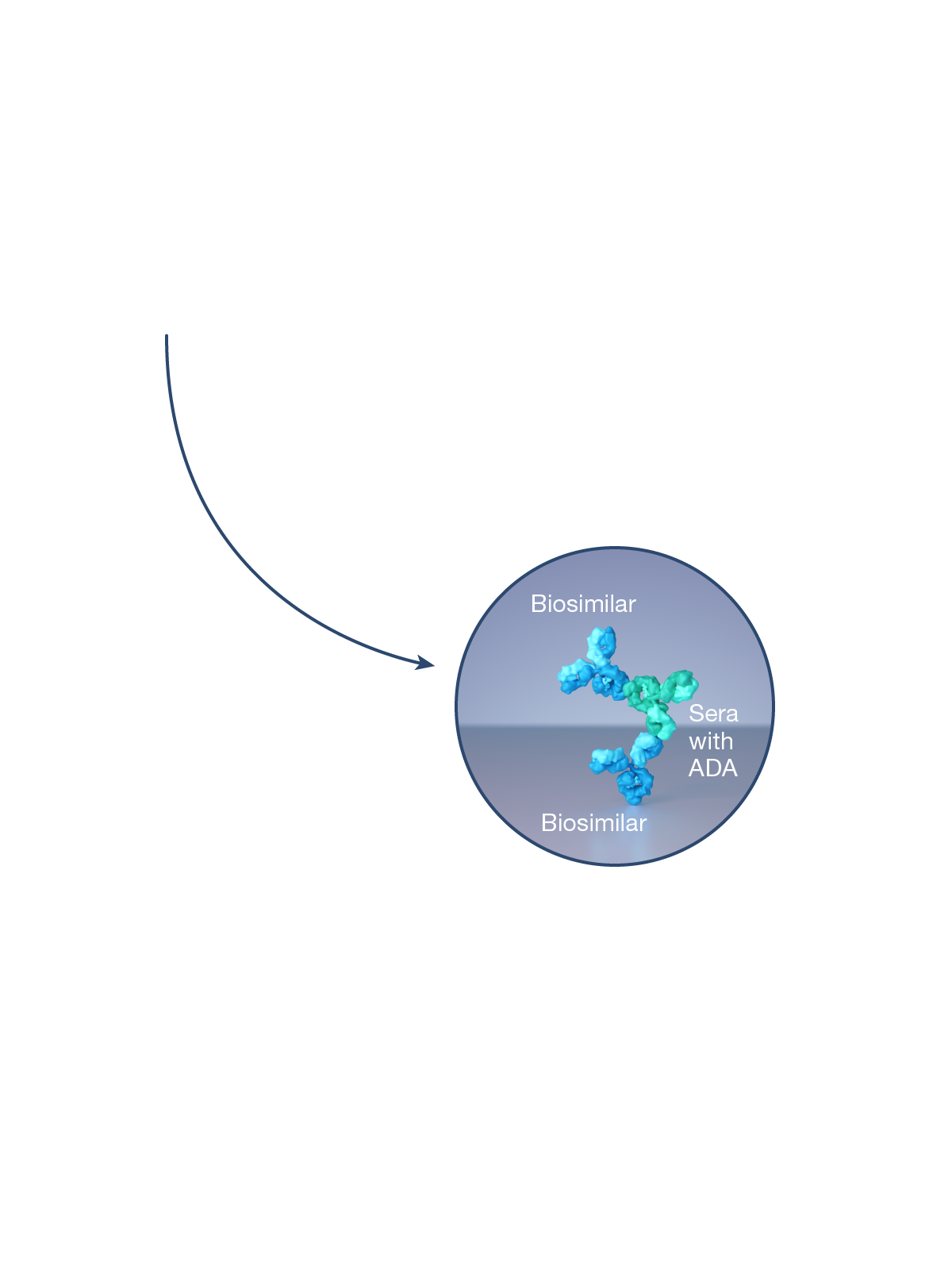
CQAs are characteristics that affect structure, purity, biological activity and stability of the biological drug.5 The development and manufacturing of biologics is a complex process, and a biosimilar has in-process quality tests during manufacturing that typically number in the hundreds.6 Each manufacturer determines the extent of testing and discusses their plan with health authorities.1































 References
References